




SUPLEMENTO ONCOLOGÍA - LEUCEMIA
Marcadores moleculares:
Su importancia en la leucemia mieloide aguda

Alexis M. Cruz Chacón, MD FACP
Hematólogo Oncólogo
Especialista en Trasplante de Médula Ósea en Adultos
Director Médico, Programa de Trasplante de Médula Ósea
Hospital Auxilio Mutuo
Sección Hematología y Oncología, Hospital Municipal de San Juan
Clínica Malignidades Hematológicas, Hospital Centro Comprensivo de Cáncer UPR
La leucemia mieloide aguda (AML) es un tipo de cáncer que afecta las células de la sangre y es el tipo de leucemia aguda que se observa más frecuentemente en los pacientes adultos.
Incidencia en Puerto Rico
La leucemia mieloide aguda es el tercer cáncer hematológico más común en Puerto Rico. Según las más recientes estadísticas publicadas por el Registro de Cáncer de la Universidad de Puerto Rico, se reportaron un total de 640 casos de leucemia mieloide aguda en la isla durante el periodo entre 2016 y 2019. Los municipios con la mayor incidencia de leucemias se encuentran en la región sur de Puerto Rico
Diagnóstico
Para hacer un diagnóstico claro y definitivo de una leucemia, es necesario realizar y analizar pruebas de sangre y una biopsia de médula ósea. Además de los análisis de patología, se realizan pruebas genéticas para poder identificar anormalidades en los cromosomas que contribuyen al diagnóstico, a su clasificación y al mejor tratamiento de la enfermedad.
Con el paso de los años, el surgimiento de nuevas tecnologías ha dado paso a la identificación de marcadores moleculares, como las mutaciones genéticas, que están también asociados a la enfermedad. Estos marcadores moleculares han contribuido a entender mejor los distintos mecanismos que causan la enfermedad y se han vuelto esenciales para el monitoreo, el diagnóstico, el pronóstico y el tratamiento de la AML.
En la actualidad, junto con las anormalidades citogenéticas, los marcadores moleculares asociados a la AML son considerados factores pronósticos importantes que están relacionados con los rasgos clínicos, con la respuesta terapéutica, con la tasa de recurrencia y con la supervivencia.
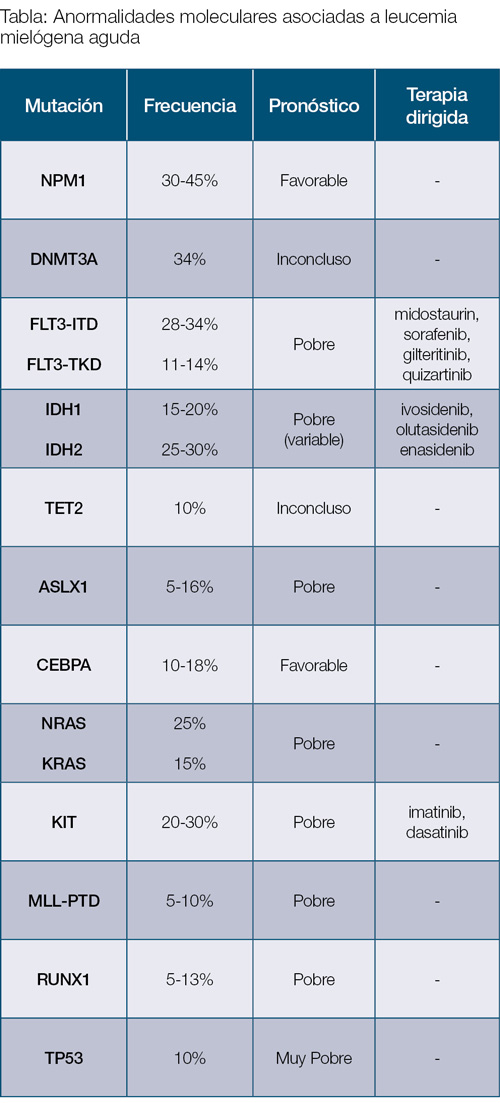
Principales marcadores moleculares para la leucemia mieloide aguda (AML)
En este artículo, se resumen y discuten algunos de los marcadores moleculares más importantes en la AML, así como su relevancia clínica y terapéutica.
Nucleophosmin 1 (NPM1):
La mutación en el gen NPM1 es la mutación que se asocia más comúnmente a la AML y se encuentra en un 40% a un 60% de los pacientes con AML que han tenido un estudio de citogenética normal. El gen de NPM1 se encuentra en el cromosoma 5 y codifica proteínas que se transportan entre en núcleo y el citoplasma de la célula y que están envueltas en algunos procesos como la reparación del ADN y la estabilización de los genes supresores de tumor.
Valor pronóstico:
Desde el punto de vista práctico, es importante considerar que la presencia de la mutación en NPM1 de forma aislada –sin otras mutaciones de alto riesgo que la acompañen– está asociada a un buen pronóstico. Por eso, en estos pacientes no se recomienda un trasplante alogénico de médula ósea como parte del tratamiento luego de recibir quimioterapia y de alcanzar una remisión completa.
Gen FLT3 (Fms-like tyrosine kinase 3):
El gen FLT3 está localizado en el cromosoma-13 y codifica para un receptor de quinasas de tirosina que participa en el proceso de proliferación, de diferenciación y de apoptosis de las células progenitoras hematopoyéticas.
La mutación en FLT3 se encuentra en un 30% de los pacientes de AML y puede ocurrir en dos variantes: FLT3-ITD -mutaciones de duplicación del tándem interno- (en un 20 a 25%) y FLT3-TKD -mutaciones en el dominio de tirosina quinasa- (en un 5 a 10% de los pacientes con AML recién diagnosticados).
Valor pronóstico:
Es importante mencionar que las mutaciones en FLT3 –en especial aquellas en la variante FLT3-ITD– en pacientes con AML están asociadas a un pronóstico más desfavorable debido a una mayor tasa de recaída y a una menor supervivencia global. Los pacientes con AML con mutación de FLT3 se caracterizan por presentar conteos bien elevados de glóbulos blancos y células inmaduras en la sangre.
Tratamiento:
Desde el punto de vista de los tratamientos, la realidad de que la mutación de FLT3 sea una de las que más se ha estudiado y que haya sido más utilizada como un objetivo terapéutico ha dado paso a que tengamos disponibles en la actualidad varias terapias dirigidas que inhiben FLT3. Esto permite eliminar más efectivamente las células de leucemia.
Actualmente, existen 3 inhibidores de FLT3 aprobados: midostaurin, gilteritinib y, recientemente, quizartinib. La integración de estas terapias dirigidas al plan de tratamiento para los pacientes con AML ha permitido mejorar significativamente la respuesta terapéutica en comparación con la quimioterapia tradicional sola.
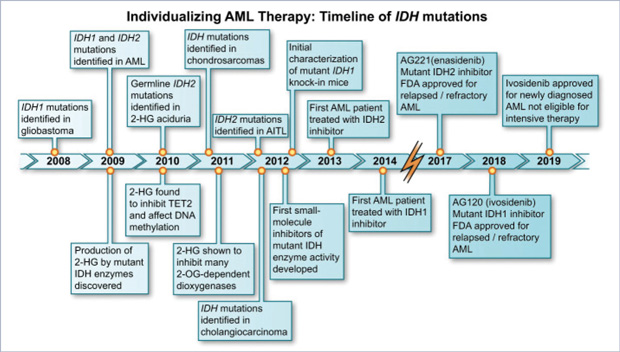
Gen IDH1/IDH2:
Los genes IDH1 e IDH2 se encuentran en los cromosomas 2 y 15 respectivamente, y codifican para unas proteínas supresoras de tumor, que están también involucradas en el metabolismo, y en la producción de energía del organismo. Las mutaciones en IDH1/2 causan un arresto en el proceso de diferenciación normal de la médula ósea.
Estas mutaciones se encuentran en un 15% a un 20% (la mutación IDH1) y en un 25% a un 30% (la mutación IDH2) de los pacientes con AML. La presentación más común de estos pacientes con mutaciones de IDH1/2 es con conteos bajos de células blancas y conteo de plaquetas elevado.
Valor pronóstico:
La presencia de mutaciones de IDH1/2 en pacientes de AML está asociada a un pronóstico pobre. Ambas mutaciones han demostrado ser un marcador confiable de enfermedad residual, el cual puede ayudar a predecir o identificar una recaída a tiempo.
Tratamiento:
Los genes IDH1 e IDH2 también han sido utilizadas como objetivos terapéuticos. Desde este punto de vista –terapéutico–, ya existen actualmente 2 inhibidores de IDH1 (ivosidenib, olutasidenib) y 1 inhibidor de IDH2 (enasidenib) que están aprobados por la FDA para el tratamiento de la AML.
Gen TP53:
El gen de TP53 se localiza en el cromosoma 17 y codifica un factor de transcripción clave involucrado en el proceso de reparación del ADN. Una mutación en este gen supresor de tumor provoca un descontrol en la proliferación celular, dando paso a la creación y desarrollo de un tumor. Las mutaciones del gen TP53 existen en una amplia variedad de tipos de cáncer que incluyen cáncer de ovario, cáncer de esófago, cáncer colorrectal y cáncer de pulmón. La mutación de TP53 se encuentra en el 10% de los casos nuevos de AML, en el 20% al 37% de casos de AML relacionados con el tratamiento o de AML secundaria, y en el 70% de los casos de AML con cariotipo complejo. La mutación de TP53 es más común en pacientes de edad avanzada y en aquellos con anormalidades de los cromosomas 5 y 7.
Valor pronóstico:
Se trata de un marcador de muy pobre pronóstico que está asociado a baja respuesta terapéutica, a alta recurrencia y a una supervivencia más corta.
Tratamiento:
Actualmente, las opciones de tratamiento para pacientes con AML con mutación de TP53 son muy limitadas. Las terapias existentes, como la quimioterapia y el trasplante alogénico de médula ósea, no han demostrado ser tan efectivas. Se piensa que esto ocurre debido al desarrollo de resistencia a agentes de quimioterapia. Se están realizando múltiples estudios para conseguir mejores tratamientos en este grupo de pacientes, muchos de ellos en el campo de la inmunoterapia y la terapia celular. Por lo tanto, es altamente recomendado ingresar en estudios clínicos a pacientes con AML con mutación de TP53.
Comentarios
El descubrimiento y la caracterización de anormalidades citogenéticas y moleculares específicas asociadas a los distintos tipos de leucemia han ayudado a definir mejor el pronóstico y la sobrevida de estas condiciones. Esto ha permitido desarrollar sistemas de clasificación y asignación de riesgo asociado a la enfermedad, que nos vienen ofreciendo mejores herramientas para poder tomar decisiones clínicas y terapéuticas de una manera más segura y certera.
De igual forma, el descubrimiento de mutaciones específicas asociadas a la AML es lo que ha dado paso al desarrollo de nuevas terapias biológicas para el tratamiento de esta enfermedad en los últimos 10 años. Los tratamientos conocidos como agentes biológicos o terapias dirigidas tienen la capacidad de atacar moléculas y mutaciones específicas de las que dependen las células cancerosas para sobrevivir. Este mecanismo de acción hace que estén más dirigidos a la célula maligna y que causen menos daño a las células normales en el paciente, lo que resulta en menor toxicidad y mayor tolerancia.
Pese al sólido avance que se ha dado en el tratamiento de leucemia en la última década, es importante fomentar el desarrollo de estudios clínicos para nuestros pacientes en Puerto Rico, en especial para aquellos que no responden a los tratamientos disponibles. Desde ese punto de vista, es válido mencionar que todas las terapias emergentes surgieron gracias a este tipo de investigaciones.
Referencias
- Issa GC, DiNardo CD. Acute myeloid leukemia with IDH1 and IDH2 mutations: 2021 treatment algorithm. Blood Cancer J. 11, 107 (2021). https://doi.org/10.1038/s41408-021-00497-1.
- Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066 (2009).
- DiNardo CD, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am. J. Hematol. 90, 732–736 (2015).
- Molenaar RJ, et al. Clinical and biological implications of ancestral and non-ancestral IDH1 and IDH2 mutations in myeloid neoplasms. Leukemia 29, 2134–2142 (2015).
- Ward PS, et al. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J Biol Chem. 288, 3804–3815 (2013).
- Dang L, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 462, 739–744 (2009).
- Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016 Jun 9; 374(23): 2209-2221. doi: 10.1056/NEJMoa1516192. PMID: 27276561
- Hunter AM, Sallman DA. Current status and new treatment approaches in TP53 mutated AML. Best Pract Res Clin Haematol. 2019 Jun;32(2):134-144. doi: 10.1016/j.beha.2019.05.004.
- Weinberg OK, Siddon A, Madanat YF, et al. TP53 mutation defines a unique subgroup within complex karyotype de novo and therapy-related MDS/AML. Blood Adv. 2022 May 10;6(9):2847-2853. doi: 1182/bloodadvances.2021006239. PMID: 35073573
- Weinberg OK, Porwit A, Orazi A, et al. The International Consensus Classification of acute myeloid leukemia. Virchows Arch. 2023 Jan;482(1): 27-37. doi: 10.1007/s00428-022-03430-4.
- George B, Kantarjian H, Baran N, et al. TP53 in Acute Myeloid Leukemia: Molecular Aspects and Patterns of Mutation. Int J Mol Sci. 2021 Oct 5;22(19):10782. doi: 10.3390/ijms221910782.
- Sallman DA, McLemore AF, Aldrich AL, et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood. 2020 Dec 10;136(24):2812-2823. doi: 10.1182/blood.2020006158.
- Metzeler KH, Herold T, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016 Aug 4; 128(5): 686-698. doi: 10.1182/blood-2016-01-693879.
- Welch JS, Petti AA, Miller CA, et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N Engl J Med. 2016 Nov 24; 375(21): 2023-2036. doi: 10.1056/NEJMoa1605949.
- Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med (2015) 373(12):1136–1152. doi: 10.1056/NEJMra1406184
- Carow CE, Levenstein M, et al. Expression of the hematopoietic growth factor receptor FLT3 in human leukemias. Blood (1996) 87(3):1089–1096. doi: 10.1182/blood.V87.3.1089.bloodjournal8731089
- Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood (2002) 100(5):1532–1542.
- Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med (2016) 374(23):2209–21. doi: 10.1056/NEJMoa1516192
- Schnittger S, Bacher U, Haferlach C, et al. Diversity of the juxtamembrane and TKD1 mutations (exons 13-15) in the FLT3 gene with regards to mutant load, sequence, length, localization, and correlation with biological data. Genes Chromosomes Cancer (2012) 51(10):910–24. doi: 10.1002/gcc.21975
- Dohner H, Estey E, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood (2017) 129(4):424–47.
- Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood (2007) 110(4):1262–1270.
- Tallman MS, Wang ES, Altman JK, et al. AML, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw (2019) 17(6):721–49. doi: 10.6004/jnccn.2019.0028
- Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML. Leukemia (2019) 33(2):299–312.
- Levis MJ, Perl AE, Altman JK, et al. A next-generation sequencing-based assay for minimal residual disease assessment in AML patients with FLT3-ITD mutations. Blood Adv (2018) 2(8):825–831.
- Spencer DH, Abel HJ, Lockwood CM, et al. Detection of FLT3 internal tandem duplication in targeted, short-read-length, next-generation sequencing data. J Mol Diagn (2013) 15(1):81–93.
- Bolli N, Manes N, McKerrell T, et al. Characterization of gene mutations and copy number changes in AML using a rapid target enrichment protocol. Haematologica (2015) 100(2):214–222.



