


| * | Claims data 07/2017-06/2020. Source: IntrinsiQ Data © 2020, IntrinsiQ Specialty Solutions, Inc. |



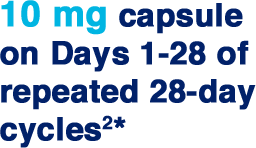
| * | If tolerated, dose can be increased to 15 mg after 3 cycles. Starting dose adjustments are needed for renal impairment. |

WARNING: EMBRYO-FETAL TOXICITY, HEMATOLOGIC TOXICITY, and VENOUS and ARTERIAL THROMBOEMBOLISM.
Indications and Important Safety Info
REVLIMID® (lenalidomide) is indicated as maintenance therapy in adult patients with MM following autologous hematopoietic stem cell transplantation (auto‑HSCT).
REVLIMID is not indicated and is not recommended for the treatment of patients with chronic lymphocytic leukemia (CLL) outside of controlled clinical trials.
WARNING: EMBRYO-FETAL TOXICITY, HEMATOLOGIC TOXICITY, and VENOUS and ARTERIAL THROMBOEMBOLISM
Embryo-Fetal Toxicity
Do not use REVLIMID during pregnancy. Lenalidomide, a thalidomide analogue, caused limb abnormalities in a developmental monkey study. Thalidomide is a known human teratogen that causes severe life-threatening human birth defects. If lenalidomide is used during pregnancy, it may cause birth defects or embryo‑fetal death. In females of reproductive potential, obtain 2 negative pregnancy tests before starting REVLIMID treatment. Females of reproductive potential must use 2 forms of contraception or continuously abstain from heterosexual sex during and for 4 weeks after REVLIMID treatment. To avoid embryo-fetal exposure to lenalidomide, REVLIMID is only available through a restricted distribution program, the REVLIMID REMS® program.
Information about the REVLIMID REMS program is available at www.celgeneriskmanagement.com or by calling the manufacturer’s toll‑free number 1‑888‑423‑5436.
Hematologic Toxicity (Neutropenia and Thrombocytopenia)
REVLIMID can cause significant neutropenia and thrombocytopenia. Eighty percent of patients with del 5q MDS had to have a dose delay/reduction during the major study. Thirty‑four percent of patients had to have a second dose delay/reduction. Grade 3 or 4 hematologic toxicity was seen in 80% of patients enrolled in the study. Patients on therapy for del 5q MDS should have their complete blood counts monitored weekly for the first 8 weeks of therapy and at least monthly thereafter. Patients may require dose interruption and/or reduction. Patients may require use of blood product support and/or growth factors.
Venous and Arterial Thromboembolism
REVLIMID has demonstrated a significantly increased risk of deep vein thrombosis (DVT) and pulmonary embolism (PE), as well as risk of myocardial infarction and stroke in patients with MM who were treated with REVLIMID and dexamethasone therapy. Monitor for and advise patients about signs and symptoms of thromboembolism. Advise patients to seek immediate medical care if they develop symptoms such as shortness of breath, chest pain, or arm or leg swelling. Thromboprophylaxis is recommended and the choice of regimen should be based on an assessment of the patient’s underlying risks.
CONTRAINDICATIONS
Pregnancy: REVLIMID can cause fetal harm when administered to a pregnant female and is contraindicated in females who are pregnant. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential risk to the fetus.
Severe Hypersensitivity Reactions: REVLIMID is contraindicated in patients who have demonstrated severe hypersensitivity (e.g., angioedema, Stevens-Johnson syndrome, toxic epidermal necrolysis) to lenalidomide.
WARNINGS AND PRECAUTIONS
Embryo-Fetal Toxicity: See Boxed WARNINGS
- Females of Reproductive Potential: See Boxed WARNINGS.
- Males: Lenalidomide is present in the semen of patients receiving the drug. Males must always use a latex or synthetic condom during any sexual contact with females of reproductive potential while taking REVLIMID and for up to 4 weeks after discontinuing REVLIMID, even if they have undergone a successful vasectomy. Male patients taking REVLIMID must not donate sperm.
- Blood Donation: Patients must not donate blood during treatment with REVLIMID and for 4 weeks following discontinuation of the drug because the blood might be given to a pregnant female patient whose fetus must not be exposed to REVLIMID.
REVLIMID REMS® Program: See Boxed WARNINGS: Prescribers and pharmacies must be certified with the REVLIMID REMS program by enrolling and complying with the REMS requirements; pharmacies must only dispense to patients who are authorized to receive REVLIMID. Patients must sign a Patient-Physician Agreement Form and comply with REMS requirements; female patients of reproductive potential who are not pregnant must comply with the pregnancy testing and contraception requirements and males must comply with contraception requirements.
Hematologic Toxicity: REVLIMID can cause significant neutropenia and thrombocytopenia. Monitor patients with neutropenia for signs of infection. Advise patients to observe for bleeding or bruising, especially with use of concomitant medications that may increase risk of bleeding. Patients may require a dose interruption and/or dose reduction. MM: Monitor complete blood counts in patients taking REVLIMID + dexamethasone or REVLIMID as maintenance therapy, every 7 days for the first 2 cycles, on days 1 and 15 of cycle 3, and every 28 days thereafter.
Venous and Arterial Thromboembolism: See Boxed WARNINGS: Venous thromboembolic events (DVT and PE) and arterial thromboses (MI and CVA) are increased in patients treated with REVLIMID. Patients with known risk factors, including prior thrombosis, may be at greater risk and actions should be taken to try to minimize all modifiable factors (e.g., hyperlipidemia, hypertension, smoking). Thromboprophylaxis is recommended and the regimen should be based on the patient’s underlying risks. Erythropoietin-stimulating agents (ESAs) and estrogens may further increase the risk of thrombosis and their use should be based on a benefit-risk decision.
Increased Mortality in Patients With CLL: In a clinical trial in the first-line treatment of patients with CLL, single-agent REVLIMID therapy increased the risk of death as compared to single‑agent chlorambucil. Serious adverse cardiovascular reactions, including atrial fibrillation, myocardial infarction, and cardiac failure, occurred more frequently in the REVLIMID arm. REVLIMID is not indicated and not recommended for use in CLL outside of controlled clinical trials.
Second Primary Malignancies (SPM): In clinical trials in patients with MM receiving REVLIMID and in patients with FL or MZL receiving REVLIMID + rituximab therapy, an increase of hematologic plus solid tumor SPM, notably AML, have been observed. In patients with MM, MDS was also observed. Monitor patients for the development of SPM. Take into account both the potential benefit of REVLIMID and risk of SPM when considering treatment.
Increased Mortality With Pembrolizumab: In clinical trials in patients with MM, the addition of pembrolizumab to a thalidomide analogue plus dexamethasone resulted in increased mortality. Treatment of patients with MM with a PD‑1 or PD‑L1 blocking antibody in combination with a thalidomide analogue plus dexamethasone is not recommended outside of controlled clinical trials.
Hepatotoxicity: Hepatic failure, including fatal cases, has occurred in patients treated with REVLIMID + dexamethasone. Pre-existing viral liver disease, elevated baseline liver enzymes, and concomitant medications may be risk factors. Monitor liver enzymes periodically. Stop REVLIMID upon elevation of liver enzymes. After return to baseline values, treatment at a lower dose may be considered.
Severe Cutaneous Reactions: Severe cutaneous reactions including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reaction with eosinophilia and systemic symptoms (DRESS) have been reported. These events can be fatal. Patients with a prior history of Grade 4 rash associated with thalidomide treatment should not receive REVLIMID. Consider REVLIMID interruption or discontinuation for Grade 2‑3 skin rash. Permanently discontinue REVLIMID for Grade 4 rash, exfoliative or bullous rash, or for other severe cutaneous reactions such as SJS, TEN, or DRESS.
Tumor Lysis Syndrome (TLS): Fatal instances of TLS have been reported during treatment with REVLIMID. The patients at risk of TLS are those with high tumor burden prior to treatment. Closely monitor patients at risk and take appropriate preventive approaches.
Tumor Flare Reaction (TFR): TFR has occurred during investigational use of REVLIMID for CLL and lymphoma. Monitoring and evaluation for TFR is recommended in patients with MCL, FL, or MZL. Tumor flare may mimic the progression of disease (PD). In patients with Grade 3 or 4 TFR, it is recommended to withhold treatment with REVLIMID until TFR resolves to ≤Grade 1. REVLIMID may be continued in patients with Grade 1 and 2 TFR without interruption or modification, at the physician’s discretion.
Impaired Stem Cell Mobilization: A decrease in the number of CD34+ cells collected after treatment (>4 cycles) with REVLIMID has been reported. Consider early referral to transplant center to optimize timing of the stem cell collection.
Thyroid Disorders: Both hypothyroidism and hyperthyroidism have been reported. Measure thyroid function before starting REVLIMID treatment and during therapy.
Early Mortality in Patients With MCL: In another MCL study, there was an increase in early deaths (within 20 weeks); 12.9% in the REVLIMID arm versus 7.1% in the control arm. Risk factors for early deaths include high tumor burden, MIPI score at diagnosis, and high WBC at baseline (≥10 x 109/L).
Hypersensitivity: Hypersensitivity including angioedema, anaphylaxis, and anaphylactic reactions to REVLIMID has been reported. Permanently discontinue REVLIMID for these reactions.
ADVERSE REACTIONS
Multiple Myeloma
- Maintenance Therapy Post Auto-HSCT: The most frequently reported Grade 3 or 4 reactions in ≥20% (REVLIMID arm) included neutropenia, thrombocytopenia, and leukopenia. The serious adverse reactions of lung infection and neutropenia (more than 4.5%) occurred in the REVLIMID arm.
- The most frequently reported adverse reactions in ≥20% (REVLIMID arm) across both maintenance studies (Study 1, Study 2) were neutropenia (79%, 61%), thrombocytopenia (72%, 24%), leukopenia (23%, 32%), anemia (21%, 9%), upper respiratory tract infection (27%, 11%), bronchitis (4%, 47%), nasopharyngitis (2%, 35%), cough (10%, 27%), gastroenteritis (0%, 23%), diarrhea (54%, 39%), rash (32%, 8%), fatigue (23%, 11%), asthenia (0%, 30%), muscle spasm (0%, 33%), and pyrexia (8%, 20%).
DRUG INTERACTIONS
Periodically monitor digoxin plasma levels due to increased Cmax and AUC with concomitant REVLIMID therapy. Patients taking concomitant therapies such as ESAs or estrogen-containing therapies may have an increased risk of thrombosis. It is not known whether there is an interaction between dexamethasone and warfarin. Close monitoring of PT and INR is recommended in patients with MM taking concomitant warfarin.
USE IN SPECIFIC POPULATIONS
- PREGNANCY: See Boxed WARNINGS: If pregnancy does occur during treatment, immediately discontinue the drug and refer patient to an obstetrician/gynecologist experienced in reproductive toxicity for further evaluation and counseling. There is a REVLIMID pregnancy exposure registry that monitors pregnancy outcomes in females exposed to REVLIMID during pregnancy as well as female partners of male patients who are exposed to REVLIMID. This registry is also used to understand the root cause for the pregnancy. Report any suspected fetal exposure to REVLIMID to the FDA via the MedWatch program at 1‑800‑FDA‑1088 and also to Celgene Corporation at 1‑888‑423‑5436.
- LACTATION: There is no information regarding the presence of lenalidomide in human milk, the effects of REVLIMID on the breastfed infant, or the effects of REVLIMID on milk production. Because many drugs are excreted in human milk and because of the potential for adverse reactions in breastfed infants from REVLIMID, advise female patients not to breastfeed during treatment with REVLIMID.
- RENAL IMPAIRMENT: Adjust the starting dose of REVLIMID based on the creatinine clearance value and for patients on dialysis.
Important Dosing Information
- The capsules should not be opened, broken, or chewed
- REVLIMID is primarily excreted unchanged by the kidney. Since elderly patients are more likely to have decreased renal function, care should be taken in dose selection. Monitor renal function
- Monitor CBCs every 7 days (weekly) for the first 2 cycles, on Days 1 and 15 of Cycle 3, and every 28 days (4 weeks) thereafter
- Treatment is continued or modified based on clinical and laboratory findings
- Dose modification guidelines are recommended to manage Grade 3/4 neutropenia or thrombocytopenia. For non-hematologic Grade 3/4 toxicities judged to be related to REVLIMID, hold treatment and restart at the physician’s discretion at next lower dose level when toxicity has resolved to Grade 2 or below
- Patients may require dose interruption and/or reduction
- Patients may require the use of blood product support and/or growth factors
Please see full Prescribing Information, including Boxed WARNINGS, for REVLIMID.
auto-HSCT, autologous hematopoietic stem cell transplantation; CBC, complete blood count; MM, multiple myeloma.
REFERENCES: 1. Data on file. Bristol-Myers Squibb Co; 2020. 2. REVLIMID [package insert]. Summit, NJ: Celgene Corp.