- Galenus 30

- Carta del Editor
- Editorial Invitado
- Reconocimiento
- La futura Doctora
- Articulos Médicos
- Laboratorio
- Suplemento Cirugía Vascular y Endovascular
- Suplemento de Endocrinología y Diabetología
- Suplemento Radiología
- Suplemento Reumatología
- Historia
- Léxico Médico
- Institucionales
- Eventos y Actividades
- Temas de interés
- Edición impresa


Cuidado médico del paciente pediátrico hemofílico en Puerto Rico

Pedro J. Santiago-Borrero, MD
Hematólogo
Decano de la Escuela de Medicina,
Recinto de Ciencias Médicas
Universidad de Puerto Rico
Etiología e incidencia
La hemofilia es un desorden genético ligado al cromosoma X, por lo que prácticamente solo se expresa en varones. Su incidencia es parecida en todos los países del mundo y es de 1 por cada 5000 o 6000 varones.
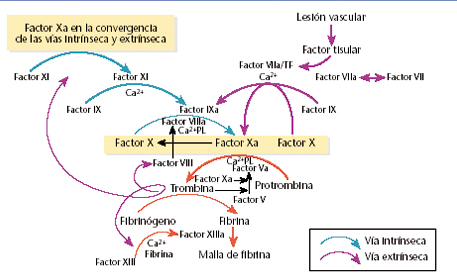
La hemofilia A (clásica) es causada por deficiencia del factor VIII de coagulación y representa a cerca del 80% de todos los hemofílicos.
Otra enfermedad relacionada es la hemofilia B, causada por deficiencia del factor IX. La presentación clínica de estas dos enfermedades es indistinguible. Aún no hay una cura para esta enfermedad, pero sí se han logrado respuestas parciales y por periodos cortos de tiempo con trasplante de hígado.
Hemofilia en Puerto Rico
Primera mitad del siglo XX _ Desde el comienzo del siglo XX se sospechaba la existencia de hemofilia en Puerto Rico. A finales de la década de 1950, algunos de nuestros médicos comenzaron a identificar a estos pacientes y a tratarlos con plasma humano normal, que ya estaba disponible en nuestros bancos de sangre. En esa época, ya se sabía que muchos de los pacientes con una enfermedad parecida a la hemofilia residían o descendían de familias que vivían en la región central de la isla de Puerto Rico.
La calidad del cuidado a los pacientes hemofílicos en Puerto Rico comenzó a mejorar en la década de 1960, cuando se hizo más disponible el uso de plasma y del crioprecipitado de dicho plasma en nuestros bancos de sangre.
La década de 1970 _ El siguiente cambio significativo ocurrió a comienzos de la década de 1970, cuando nuestros laboratorios ya nos permitían separar correctamente los pacientes de hemofilia A de aquellos con hemofilia B o con otros desórdenes de coagulación. En esa década, ya nuestros hospitales comenzaban a adquirir los concentrados de factores VIII y IX, extraídos de plasma humano normal.
Lamentablemente, en la misma década, la preparación de concentrados de factores de coagulación tuvo graves problemas de contaminación con el virus de inmunodeficiencia humana (VIH). Casi el 50% de nuestros pacientes hemofílicos adquirieron dicha enfermedad y murieron en una o dos décadas.
Décadas de 1980 y 1990 _ En las décadas de 1980 y 1990, ocurrieron dos eventos significativos para el cuidado a nuestros pacientes hemofílicos. Uno de ellos fue el rápido desarrollo de la tecnología en la producción de factor VIII, así como de otros factores de coagulación (no derivados de plasma), usando las técnicas recombinantes libres de agentes infecciosos. Nuestro hospital fue muy generoso en la compra de esos factores.
El otro logro importante de esa época fue el establecimiento del Centro Comprensivo de Hemofilia de Puerto Rico (CCHPR) en el Hospital Pediátrico Universitario. Esto se logró mediante una propuesta financiada por el Departamento de Salud de Estados Unidos (MCHB, HHS) y la cooperación de dos reconocidos hematólogos, el Dr. Louis Aledort y la Dra. Margaret Hillgartner, directores de los Centros de Hemofilia del Centro Médico de Mount Sinai y del Centro Médico de la Universidad de Nueva York, respectivamente, en Nueva York.
Con dichos recursos y el correspondiente respaldo de la administración del Hospital Pediátrico y del Departamento de Salud de Puerto Rico, se ha desarrollado un excelente programa de cuidado integral a niños, adolescentes y algunos adultos jóvenes, durante los últimos 28 años. Actualmente, más del 95% de los niños y adolescentes con hemofilia en Puerto Rico reciben tratamiento integral y continuo en nuestro CCHPR.
Manifestaciones clínicas de la hemofilia
La manifestación principal de la hemofilia es el sangrado excesivo causado por trauma, inclusive leve, fuera de lo esperado en una persona normal. A veces, el sangrado puede ocurrir espontáneamente o con la actividad usual de la vida diaria.
Los lugares de sangrado más frecuentes e incapacitantes en estos pacientes son las articulaciones, músculos y piel, además del tracto intestinal y urinario, la cavidad oral y nasal; y dentro de órganos internos, el cerebro, pulmón, hígado, bazo, intestinos o riñones, entre otros.
Las manifestaciones clínicas de la hemofilia se dividen en tres niveles de severidad: severo (menos del 1%); moderado (de 1 a 5%) y leve (más de 5%).
Genética molecular de la hemofilia
Las hemofilias A y B, causadas por deficiencia de los factores de coagulación VIII y IX, respectivamente, son los desórdenes congénitos de coagulación más comunes, después de la enfermedad de Von Willebrand. Los genes de esas dos enfermedades se localizan en el cromosoma X y, por lo tanto, se manifiestan casi exclusivamente, solo en varones.
Las mutaciones causantes de la hemofilia A y B se encuentran en los genes de los factores VIII y IX, respectivamente. El gen de factor VIII es bastante grande, con longitud de 186 kilobases, lo que explica su mayor riesgo de mutaciones y, por ende, una mayor prevalencia en comparación con la de factor IX, cuyo gen mide solo 34 kilobases. Las alteraciones mayores de estos genes causan enfermedades severas, en las que se han descubierto más de 600 mutaciones.
Características clínicas de la hemofilia
La información más útil en el diagnóstico de la hemofilia se obtiene mediante el historial médico, el examen físico, las características del sangrado y las complicaciones de la enfermedad.
Los desórdenes hemostáticos secundarios, que son causados por alguna anormalidad en la propagación de las reacciones de coagulación, se caracterizan principalmente por hemartrosis y sangrado intramuscular; mientras que el fallo en el componente de hemostasis primaria usualmente se manifiesta por púrpura, sangrado prolongado de heridas superficiales, epistaxis, sangrado intestinal y menorragia.
Es más probable que el sangrado recurrente de un sólo punto anatómico, como en epistaxis, sea causada por una anormalidad estructural, en vez de ser generado por un defecto hemostático.
Los síntomas de sangrado que ocurren durante un largo periodo de tiempo, especialmente desde la infancia, sugieren la presencia de un desorden hemorrágico hereditario, como la hemofilia, mientras que aquellos con manifestaciones recientes son más sugestivos de desórdenes adquiridos, principalmente relacionada con la hemostasis primaria.
Muy pocos hemofílicos sangran en el periodo neonatal. Los niños con hemofilia severa usualmente experimentan su primer sangrado a la edad de 6 a 9 meses, cuando empiezan a aumentar su movilidad, desarrollando muchos hematomas y hemartrosis, y cuando reciben inyecciones intramusculares o se someten a cirugía.
Los lugares de mayor sangrado en el niño hemofílico son las articulaciones. Esto puede ser causado por trauma, pero también puede ocurrir espontáneamente. La sangre dentro de la cavidad articular causa una reacción inflamatoria severa que, a su vez, produce hipertrofia e inflamación en el tejido sinovial. Esto causa más hemorragia y, eventualmente, aparece atrofia muscular alrededor de la articulación. Sin el tratamiento apropiado, el paciente hemofílico desarrolla artrosis degenerativa progresiva.
Los hematomas intramusculares también son característicos de la hemofilia. Los músculos grandes que sostienen mucho peso suelen ser los más afectados. El sangrado intracraneal es poco común, pero es muy serio cuando ocurre.
Diagnóstico de hemofilia
El diagnóstico de hemofilia y otras enfermedades relacionadas se sospecha mediante el historial personal y familiar, los hallazgos en el examen físico del paciente y los resultados de las pruebas de laboratorio correspondientes.
Estas pruebas incluyen inicialmente el hemograma (CBC) y el examen de morfología de las células sanguíneas, con énfasis en el número y morfología de las plaquetas, el tiempo de sangría (BT), el tiempo de protombina (PT), el tiempo parcial de tromboplastina (PTT) y la concentración de fibrinógeno.
Si los resultados de las pruebas arriba mencionadas apuntan a una probable deficiencia de uno o más factores de coagulación, se deben hacer pruebas específicas para la detección de factores deficientes, según sea la condición.
En muchos casos, con la información obtenida inicialmente, se puede establecer un diagnóstico tentativo y procederse con la administración del material o factor que sospechamos esté deficiente. Con la respuesta clínica, podemos confirmar el probable diagnóstico.
Tratamiento de la Hemofilia
En cuanto comienza un sangrado significativo en un paciente hemofílico, es necesario administrar rápido el factor deficiente (factor VIII o IX), para detener la hemorragia; aplicar inmovilización y frío a la articulación o área afectada, dar un analgésico apropiado; y administrar sangre y/o células rojas, según sea necesario.
En casos de sangrado severo, es necesario contar con la participación de especialistas, como cirujanos, ortopedas, neurólogos y especialistas de medicina física y rehabilitación, entre otros.
La dosis del factor VIII o IX para hemofilia A y B, respectivamente, varía según el lugar y la severidad del sangrado.
En un paciente con hemofilia A y sangrado leve en un lugar de poco riesgo, se administra factor VIII 20 a 25 u/k, I.V., seguido por una dosis similar cada 10 a 12 horas.
En casos de cirugía, trauma severo o sangrado intracerebral, la dosis deseable de factor VIII es de 80 a 100 u/k, con dosis subsiguientes de 40 a 50 u/k cada 10 a 12 hrs; por 7 a 8 días, dependiendo de la severidad y localización del trauma.
El manejo de pacientes con hemofilia B es similar al anterior, excepto que se administra factor IX en dosis alrededor de 1½ veces más altas que en el caso de hemofilia A.
En hemofilia A, leve y moderada, se puede usar desmopresina 0,3 ug/k s.c. cada 24 horas, con lo cual el factor VIII, usualmente, sube a niveles normales. Estos pacientes deben ser probados primero para confirmar que tienen una buena respuesta a esta medicina.
Referencias [[Textbook of Hemophilia, Volume 1. Christine A. Lee, Erik E. Berntorp & W. Keith Hoots. Blackwell Pub. Ltd 2007.]] [[Bleeding Disorders; Fast Facts; David Green and Christopher A. Lud lam. Health Press, Oxford, 2004.]]
|
Componentes del Programa Comprensivo de Hemofilia de Puerto Rico
El programa de hemofilia, que también incluye servicios para otras enfermedades hemorrágicas, está ubicado junto al Programa de Cernimiento Neonatal de Puerto Rico, en el Hospital Pediátrico Universitario, en el Centro Médico de Puerto Rico.
Este programa provee servicios esenciales para atender adecuadamente a la población pediátrica y adultos jóvenes con enfermedades hemorrágicas hereditarias en Puerto Rico. Cuenta con un amplio laboratorio de coagulación certificado, servicio de enfermería, adiestramiento especial en el manejo de estas enfermedades, consejera genética, higienista dental, nutricionista y una trabajadora social.
Los padres reciben información y adiestramiento apropiado sobre la enfermedad, para reducir riesgos y para administrar medicamentos necesarios (factores VIII o IX, entre otros) endovenosamente, en su hogar, de acuerdo a protocolos de tratamiento existentes. También se instruye a los padres sobre factores de riesgo y prevención de accidentes.
Los pacientes asisten regularmente a nuestras clínicas externas de hemofilia y otras enfermedades relacionadas. Durante los últimos años de la adolescencia, se provee información adecuada a nuestros pacientes para que vayan asumiendo responsabilidad de su propio cuidado médico, incluyendo la administración del factor de coagulación. Además, se les provee el ajuste necesario durante los estudios universitarios y/o en su trabajo, así como en la comunidad médica de adultos, lo que requiere un proceso de adaptación, que a veces ellos no comprenden y hasta rechazan.
Durante los últimos 25 años, se ha mantenido un registro de niños y jóvenes con hemofilia y enfermedades relacionadas que han recibido tratamiento en nuestro servicio. La información revisada en 2009 indica que se han diagnosticado y tratado sobre 100 casos de hemofilia A y B, en ese periodo de tiempo, lo que indica aproximadamente 4 casos nuevos por año. Esto es poco más del 80% de todos los hemofílicos que, se espera, hayan nacido en Puerto Rico en ese periodo de tiempo.
La cantidad de casos de hemofilia A y B activos en nuestro servicio en 2011 fue 89; esto es cerca de 3,6 por año, o casi el 80% de todos los neonatos afectados en Puerto Rico.|